Алкилирование ароматических аминов. Алкилирование аминов галогеналканами. Прямое алкилирование аммиака и аминов
Амины легко реагируют с галогеналканами, образуя вторичные и третичные амины, как показано выше. На последней стадии образуются четвертичные соли аммония - четыре органических группы ковалентно связаны с азотом, положительный заряд уравновешен наличием отрицательного иона. Из четвертичных солей можно выделить четвертичные основания:
2 R 4 N + X - + Ag 2 O + H 2 O → 2 AgX↓ + 2 R 4 N + OH -
Четвертичные аммониевые основания (белые кристаллические вещества) по основности сопоставимы с NaOH, КОН.
3. Ацилирование аминов (получение амидов)
Первичные и вторичные амины реагируют с ангидридами и галогенангидридами кислот с образованием амидов:

Замещенные амиды называют как производные незамещенных амидов карбоновых кислот.
Образующаяся в ходе реакции кислота связывает эквивалентное количество непрореагировавшего амина. Такой метод становится неэкономичным, если амин трудно синтезировать или он представляет собой дорогостоящий реактив. Поэтому амины часто ацилируют по реакции Шоттен-Баумана, которая заключается во взаимодействии амина и ацилирующего агента в присутствии водного раствора едкого натра:
Взаимодействие с азотистой кислотой
Азотистая кислота HONO неустойчива, но ее водный раствор можно получить, растворив при охлаждении нитрит натрия в разбавленной кислоте, например, соляной.
Первичные алифатические амины реагируют с холодным водным раствором азотистой кислоты с образованием диазониевых солей, при разложении которых образуется смесь разнообразных продуктов:
Вторичные алифатические амины реагируют с азотистой кислотой с образованием N-нитрозоаминов желтого цвета. Эти соединения, амиды азотистой кислоты, являются очень слабыми основаниями.
При взаимодействии третичных алкиламинов с азотистой кислотой образуются сложные смеси.
Образование изонитрилов
Первичные алифатические амины образуют изонитрилы при слабом нагревании с хлороформом в присутствии концентрированного раствора щелочи:
Отдельные представители
Все амины ядовиты и являются кровяными ядами. Особенно опасны их N-нитрозопроизводные.
Метиламин применяется в производстве инсектицидов, фунгицидов, ускорителей вулканизации, поверхностно-активных веществ, красителей, ракетных топлив, растворителей.
Некоторые амины применяются как селективные растворители для извлечения урана из сернокислых растворов. Амины, обладающие запахом рыбы, используются как приманка в борьбе с полевыми грызунами.
В последние годы третичные амины и соли четвертичных аммониевых оснований получили широкое распространение в качестве катализаторов межфазного переноса в органическом синтезе.
Лекция №27. АРОМАТИЧЕСКИЕ АМИНЫ
Ароматические амины. Классификация, изомерия. Номенклатура, Способыполучения: из нитросоединений (реакция Зинина) и арилгалогенидов. Получение вторичных и третичныхаминов.
Химические свойства. Влияние бензольного кольца и заместителей в нем на основность. Реакции алкилирования и ацилирования. Основания Шиффа. Реакции первичных, вторичных и третичных аминов с азотистой кислотой. Реакции электрофильного замещения у ароматическихаминов. Особенности этой реакции. Анилин, п-толуидин, N,N-диметиламин. Способы получения, применение.
Ароматические амины могут быть первичными ArNН 2 (анилин, толуидины),вторичными Ar 2 NH (дифениламин), и третичными Ar 3 N (трифениламин), а также жирноароматическими ArN(СН 3) 2 (N,N-диметиланилин).
Лекции № 41-42
АМИНЫ
Амины можно рассматривать как производные аммиака, в котором атомы водорода замещены на углеводородные радикалы.
1. Классификация , изомерия, номенклатура
В зависимости от числа углеводородных радикалов, связанных с атомом азота,
различают первичные, вторичные и третичные амины, а также четвертичные
аммониевые соли.
|
RNH 2 |
RR / NH |
RR / R // N |
RR / R // R /// N + X - |
|
первичные амины |
вторичные амины |
третичные амины |
четвертичные аммониевые соли |
По типу гибридизации атома углерода, связанного с азотом выделяют следующие группы аминов.
К этой группе относятся алкиламины, а также алкенил- и алкиниламины, в которых кратная связь удалена от атома азота. Их объединяют под названием алифатические амины . В состав этой группы входят также циклические амины, содержащие атом азота в цикле, которые являются гетероциклическими соединениями.

К этой группе принадлежат производные алкенов с атомом азота у атома углерода, образующего двойную связь – енамины (виниламины) и амины, содержащие атом азота, связанный с ароматическим кольцом - ароматические амины (ариламины).
Названия аминов образуют, добавляя к слову амин названия связанных с атомом азота углеводородных радикалов.
В другом варианте номенклатуры за основу названия принимают название родоначальной структуры (самой длинной углеродной цепи, непосредственно связанной с атомом азота) с добавлением суффикса “амин”.

В этом случае вторичные и третичные амины называют как N-замещенные производные первичных аминов.

Если молекула содержит другие функциональные группы, обозначаемые в суффиксе, то аминогруппу обозначают префиксом “амино”.
![]()
Названия диаминов образуют от названий соответствующих двухвалентных радикалов или названия родоначальной структуры с добавлением суффикса “диамин”.

Многие ароматические амины имеют тривиальные названия.

Циклические амины называют, используя номенклатуру гетероциклических соединений или, добавляя к названию двухвалентного углеводородного радикала суффикс “имин”.

Для аминов характерна изометрия углеродного скелета, изомерия положения
аминогруппы и изомерия между первичными, вторичными и третичными аминами.
2. Алифатические амины
2.1. Методы получения.
1) Алкилирование аммиака и аминов.
Аммиак взаимодействуют с алкилгалогенидами RX и другими алкилирующими реагентами (алкилсульфатами, диалкилсульфатами) с образованием на первой стадии соли алкиламмония, которая в равновесной реакции с избытком аммиака дает алкиламин. Алкиламин далее вступает в реакцию c алкилгалогенидом с образованием продукта диалкилирования и т.д. Таким образом последовательно образуются триалкиламин и соль тетраалкиламмония.

Реакция используется в основном для синтеза третичных аминов и тетраалкиламмониевых солей, так как первичные и вторичные амины, будучи более сильными нуклеофилами, чем аммиак, реагируют далее, сами предпочтительно атакуя субстрат. Приемлемые выходы первичных аминов получают при использовании большого избытка аммиака, а вторичных аминов – при большом избытке первичного амина.
Спирты алкилируют аммиак и амины в присутствии катализаторов дегидратации (Al 2 O 3 , SiO 2) при 300-500 0 C. В этом случае также образуется смесь продуктов моно-, ди- и триалкилирования.

Метод используется для получения низших алифатических аминов в промышленности.
2) Синтез первичных аминов по Габриэлю

Алкилирование фталимида калия алкилгалогенидами с последующим щелочным гидролизом или гидразинолизом N-алкилфталимида позволяет получать первичные амины без примеси вторичных и третичных. Лучше использовать протекающий в мягких условиях гидразинолиз, приводящий к образованию не растворимого в реакционной среде циклического гидразида.
3) Восстановление азотсодержащих органических соединений.
Нитрилы при восстановлении дают первичные амины. В промышленности процесс осуществляют путем каталитического гидрирования.

В препаративных целях используют восстановление алюмогидридом лития.
![]()
Введение цианогруппы (например, путем нуклеофильного замещения) и ее восстановление – синтетический прием, позволяющий нарастить углеродную цепь на один атом С.
Амиды карбоновых кислот восстанавливаются до аминов алюмогидридом лития. Из соответствующих амидов могут быть получены первичные, вторичные и третичные амины.

Восстановление азотсодержащих производных альдегидов и кетонов – оксимов и гидразонов – дает возможность превращения карбонильных соединений в первичные амины.

Для восстановления используют каталитическое гидрирование, комплексные гидриды металлов (LiAlH 4).
Нитросоединения могут быть восстановлены до первичных аминов.
![]()
В качестве восстановителей чаще всего используют металл (Fe, Zn, Sn) и кислоту; алюмогидрид лития. В алифатическом ряду метод не находит широкого применения из-за ограниченной доступности алифатических нитросоединений по сравнению с ароматическими.
Восстановление азидов дает первичные амины.

Исходные азиды легко могут быть получены из алкилгалогенидов или сульфонатов путем нуклеофильного замещения.
4) Восстановительное аминирование карбонильных соединений .
Взаимодействие альдегидов и кетонов с аммиаком в присутствии восстановителя приводит к первичным аминам.

При использовании вместо аммиака первичного амина продуктом реакции будет вторичный амин.

Процесс протекает через промежуточное образование имина с его последующим восстановлением в амин.

Восстановительное аминирование с использованием в качестве восстановителя муравьиной кислоты называют реакцией Лейкарта-Валлаха. В качестве реагентов можно использовать формиат аммония или соответствующие соли аминов.
5) Синтез аминов путем перегруппировок.
Перегруппировка Гофмана:
RCONH 2 + Br 2 + 2NaOH ® RNH 2 + 2NaBr + CO 2 + H 2 O
Перегруппировка Курциуса:

Реакции подробно рассмотрены ранее (см. лек. №36) В результате образуются
первичные амины без примеси вторичных и третичных. При этом происходит
укорочение углеродной цепи на один атом С.
2.2. Физические свойства и строение
Алифатические амины – бесцветные вещества с неприятным запахом. Низшие амины – жидкости, хорошо растворимые в воде. По растворимости они превосходят спирты с близкой молекулярной массой. Это объясняется образованием между амином и водой водородных связей типа , прочность которых сравнительно велика в силу высокой основности атома азота. Температуры кипения и плавления у третичных аминов ниже, чем у первичных и вторичных с примерно одинаковой молекулярной массой, что связано с ассоциацией последних за счет образования межмолекулярных водородных связей.

Однако эти межмолекулярные водородные связи слабее, чем у спиртов, по
причине меньшей полярности связи N-Н по сравнению со связью О-Н. Вследствие
этого амины имеют более низкие температуры кипения, чем спирты с близкой
молекулярной массой.
Амины имеют пирамидальное строение. Величины углов R-N-R близки к
тетраэдрическому – 106-108 0 . Считается, что атом азота находится в
состоянии sp 3 -гибридизации, а четвертым лигандом является
неподелённая пара электронов (“фантом”-лиганд).
Третичные амины с разными углеводородными радикалами должны быть хиральными,
так как их молекулы не имеют плоскости симметрии. Однако за счет быстрой
пирамидальной инверсии, которая представляет собой акт рацемизации, их
невозможно выделить или зафиксировать в оптически активной форме.

Четвертичные аммониевые соли в случае разных заместителей существуют в виде пары устойчивых энантиомеров.

Спектральные характеристики.
В электронных спектрах аминов наблюдается поглощение в дальней УФ-области при 195-215 нм, что соответствует возбуждению неподеленной пары электронов азота (переход n® s* ).
В ИК-спектрах первичных и вторичных аминов наблюдаются полосы поглощения, связанные с валентными колебаниями связей N-H. Первичные амины характеризуются двумя полосами поглощения при ~3400 и ~3500 см -1 , вторичные амины – одной полосой при ~3500 см -1 .
В спектрах ПМР химический сдвиг протонов связи N-H находится в области 1-5 м.д. и значительно меняется в зависимости от концентрации, температуры и растворителя.
2.3. Химические свойства
Химическое поведение аминов определяется в основном наличием свободной пары электронов у атома азота, которая обусловливает их основные и нуклеофильные свойства. Реакции с участием связей N-H и N-C под действием оснований и нуклеофильных реагентов для аминов менее характерны.
Основные свойства
Алифатические амины являются одними из самых сильных незаряженных оснований (~ 10 - 11). Их водные растворы имеют щелочную реакцию.
R 3 N + H 2 O = R 3 NH + + OH -
С неорганическими кислотами амины образуют соли, которые в большинстве случаев хорошо растворимы в воде.
R 3 N + HX = R 3 N + X -
Основность аминов зависит от их строения и природы растворителя. Сравнение
основности в водных растворах показывает, что алкиламины являются более
сильными основаниями, чем аммиак. Вторичные амины превосходят по основности
первичные. Такой ряд основности согласуется с электронодонорным влиянием
алкильных групп (+I-эффект), которое способствует делокализации положительного
заряда в сопряженной кислоте (ионе аммония) и тем самым стабилизирует её в
большей степени, чем свободный амин. Однако это не объясняет уменьшения
основности при переходе от вторичных аминов к третичным.
|
NH 3 |
C 2 H 5 NH 2 |
(C 2 H 5) 2 NH |
(C 2 H 5) 3 N |
|
|
9,25 |
10,80 |
11,09 |
10,85 |
Вероятно, такое снижение основности связано с сольватацией. Сольватация молекулами воды триалкиламмониевого катиона затруднена присутствием трех гидрофобных алкильных групп и снижением возможности образования водородных связей.

Это предположение подтверждается тем, что в газовой фазе и в малополярных
растворителях третичные амины превосходят по основности вторичные.
Нуклеофильные свойства
а) Алкилирование
Примеры реакций алкилирования обсуждались при рассмотрении методов получения аминов.
б) Ацилирование
2RNH 2 + R / COX ® R / CONHR + RNH 3 X
2R 2 NH + R / COX ® R / CONR 2 + R 2 NH 2 X
Ацилирование аминов функциональными производными карбоновых кислот дает возможность получать вторичные и третичные амиды из первичных и вторичных аминов соответственно. Реакция подробно обсуждена ранее (лекция №36).
в) Взаимодействие с сульфонилхлоридами
Сульфонилхлориды взаимодействуют с аминами, давая сульфонамиды. Реакция с бензолсульфонилхлоридом лежит в основе пробы Гинсберга , позволяющей различать и разделять первичные, вторичные и третичные амины.
Сульфонамиды, образующиеся из первичных аминов, являются NH-кислотами и со щелочами дают растворимые в воде соли.

Вторичные амины дают сульфонамиды, которые не содержат кислого водорода и не растворяются в щелочах.

Третичные амины не реагируют.
г) Нитрозирование
Нитрозирование аминов происходит при взаимодействии с азотистой кислотой в кислой среде. Неустойчивую азотистую кислоту генерируют действием сильной кислоты на нитриты. Реакция протекает по-разному в зависимости от типа амина.
Первичные алифатические амины реагируют с образованием неустойчивых алкилдиазониевых солей, которые разлагаются с выделением газообразного азота и сложной смеси продуктов дезаминирования.
Образование солей диазония – сложный многостадийный процесс, который подробно будет рассмотрен на примере ароматических аминов.
Разложение катиона алкилдиазония приводит к образованию карбокатиона, который стабилизируется путем алкилирования присутствующих в реакционной среде нуклеофилов или путем отщепления протона с образованием алкена. Этим процессам может предшествовать изомеризация карбокатиона в энергетически более стабильный ион. Так, разложение катиона н -пропилдиазония в водном растворе наряду н-пропиловым спиртом дает изопропиловый спирт, а также продукт элиминирования - пропен.

Со вторичными аминами азотистая кислота образует нерастворимые в реакционной среде нитрозамины.
R 2 NH + NaNO 2 + HCl ® R 2 N-N=O + NaCl + H 2 O
Третичные амины в сильнокислой среде при комнатной температуре с азотистой кислотой не реагируют.
Нитрозирование аминов препаративного значения не имеет. Аналитическое значение этих реакций заключается в возможности качественно различить первичные, вторичные и третичные амины.
д) Галогенирование
Первичные и вторичные амины реагируют с гипогалогенитами с образованием N-галогенаминов.

N-галогенамины – сильные окислители и галогенирующие реагенты.
Окисление
Амины дают разнообразные продукты окисления, состав которых зависит от природы окислителя и строения амина.
Перекись водорода и надкислоты окисляют третичные амины до N-оксидов.
R 3 N + HOOH ® R 3 N + -O - + H 2 O
В случае первичных и вторичных аминов первоначально образующиеся N-оксиды перегруппировываются в производные гидроксиламина.

Такое окисление протекает сложно, так как гидроксиламины сами легко окисляются. В случае первичных аминов конечными продуктами окисления являются нитросоединения, например:
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, окисляются в нитросоединения перманганатом калия в водном ацетоне.

(R=Alk; R / =H, Alk)
Кислотные свойства
Первичные и вторичные алифатические амины являются очень слабыми NH-кислотами (pK а ~33-35). Их кислотные свойства проявляются при действии щелочных металлов или таких сильных основания, как металлоорганические соединения.

Образующиеся алкил- и диалкиламиды металлов – очень сильные основания. Диалкиламиды, содержащие вторичные или третичные алкильные радикалы (например, диизопропиламид лития), представляют интерес для органического синтеза как ненуклеофильные основания. Будучи сильными основаниями, они обладают низкой нуклеофильностью по причине стерических затруднений, возникающих при атаке электрофильных центров за исключением протона. Их используют в органическом синтезе для отрыва протона и генерирования карбанионов.
Расщепление гидроксидов тетраалкиламмония по Гофману
Гидроксиды тетраалкиламмония получают действием на галогениды оксида серебра.
2R 4 N + Br - + Ag 2 O + H 2 O ® 2R 4 N + OH - + 2AgBr
В растворах гидроксиды тетраалкиламмония полностью ионизированы и являются столь же сильными основаниями, как гидроксиды натрия и калия. При нагревании они претерпевают элиминирование с образованием алкена триалкиламина и воды.
RCH 2 CH 2 (CH 3) 3 N + OH - ® RCH=CH 2 + (CH 3) 3 N + H 2 O
При наличии в молекуле нескольких b -водородных атомов процесс протекает в направлении образования наименее замещенного алкена (по правилу Гофмана ).
Причина такой ориентации при отщеплении состоит в карбанионном характере переходного состояния, что способствует отщеплению наиболее кислого протона.

При протекании процесса по механизму Е2 с “Е1 СВ -подобным” переходным состоянием на атоме углерода возникает частичный отрицательный заряд. Переходное состояние (I), предшествующее образованию продукта по правилу Зайцева, оказывается дестабилизированным за счет +I-эффекта алкильных групп. В результате процесс преимущественно направляется через наименее дестабилизированное переходное состояние (II), ведущее к продукту элиминирования по Гофману.
3. Енамины
Енамины (виниламины) устойчивы в том случае, если при атоме азота нет атомов водорода. Такие енамины можно рассматривать как азотистые аналоги виниловых эфиров. В противном случае енамины нестабильны и перегруппировываются в имины, подобно тому, как енолы изомеризуются в карбонильные соединения.
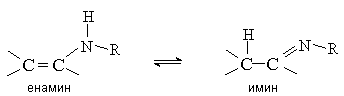
3.1. Методы получения
Основной метод получения енаминов – взаимодействие карбонильных соединений со вторичными аминами в присутствии кислотных катализаторов и средств, связывающих воду.
Реакция протекает по общему для присоединения азотистых оснований к карбонильной группе механизму.

Отщепление воды от интермедиата (III) приводит к образованию иммониевого иона (IV), который при отсутствии водорода у атома азота стабилизируется путем отщепления протона от b -углеродного атома.
3.2. Строение
Молекула енамина представляет собой р-p -сопряженную систему, строение которой можно отразить набором двух резонансных структур.
Таким образом, молекула енамина содержит два нуклеофильных центра – атом азота и атом углерода в b -положении, который несет частичный отрицательный заряд.
3.3. Химические свойства
а) Протонирование и гидролиз
Енамины являются слабыми основаниями. Их протонирование может протекать как по атому азота, так и по атому углерода. Образующаяся при протонировании по b -углеродному атому соль иммония гидролизуется, давая исходное карбонильное соединение и вторичный амин.
Гидролиз енаминов – процесс, обратный их образованию, и протекает по такому же механизму.
б) Алкилирование
Алкилирование енаминов алкилгалогенидами и другими алкилирующими реагентами протекает, как правило, по b -углеродному атому. Последующий гидролиз иммониевой соли приводит к карбонильному соединению.
Последовательность превращений – получение енамина из карбонильного соединения, алкилирование, гидролиз приводит к алкилированию исходного карбонильного соединения по a -положению и носит название реакция Сторка . Этот метод имеет преимущества перед алкилированием кетонов, так как требует более мягких условий и дает в основном продукты моноалкилирования.
Для проведения этой реакции чаще всего используют циклические амины – пирролидин, пиперидин, морфолин. Лучшие результаты достигаются при использовании активных галогенидов – аллил- и бензилгалогенидов, a -галогензамещенных производных простых и сложных эфиров. Например:

в) Ацилирование
При действии галогенангидридов и ангидридов кислот енамины дают продукты С-ацилирования. Последующий гидролиз приводит к дикарбонильному соединению.
Таким образом, последовательность реакция – получение енамина из карбонильного соединения, ацилирование, гидролиз – метод получения b -дикарбонильных соединений. Например:

4. Ароматические амины
4.1. Методы получения
1) Восстановление ароматических нитросоединений
![]()
Для восстановления в препаративных целях используют металл (Fe, Zn, Sn) и кислоту, соли металлов в низших степенях окисления (SnCl 2 , TiCl 3), сульфиды щелочных металлов, в промышленности применяют в основном каталитическое гидрирование. См. также лекцию №40.
2) Алкилирование

Реакция аналогична алкилированию алифатических аминов. В качестве алкилирующих реагентов используют алкилгалогениды, алкилсульфаты, спирты.
3) Арилирование
Галогенарены реагируют с аммиаком и аминами в жестких условиях. Процесс катализируется медью и ее соединениями.
Реакция замещения галогена протекает легче при наличии в орто - и пара -положениях электроноакцепторных групп (NO 2 , CN).
Галогенарены взаимодействуют с ариламинами в присутствии меди с образованием диариламинов (реакция Ульмана).

4.2. Физические свойства и строение
Ароматические амины – бесцветные жидкости или твердые вещества. При хранении быстро темнеют вследствие окисления кислородом воздуха.
Аминогруппа и ароматическое кольцо образуют сопряженную систему. Аминогруппа проявляет электронодонорные свойства за счет +М-эффекта.

Ароматические амины обладают сильными электронодонорными свойствами, на что указывают низкие энергии ионизации (для анилина 7,7 эВ, для фенола 8,4 эВ).
4.3. Химические свойства
Для ариламинов характерны реакции с электрофильными реагентами. Местом атаки электрофила может быть атом азота или ароматическое кольцо.
Основные свойства
Ароматические амины обладают меньшей основностью, чем алифатические амины и аммиак (~ 3 – 5). Причина низкой основности ариламинов – стабилизация свободного амина за счет делокализации неподеленной пары электронов азота по ароматическому кольцу и потеря энергии стабилизации при нарушении сопряженной системы в результате протонирования.
Дифениламин и трифениламин имеют еще большие возможности для делокализации пары электронов азота, что приводит значительному снижению основности. Трифениламин практически не обладает основными свойствами.
п-CH 3 C 6 H 4 NH 2п-O 2 NC 6 H 4 NH 2
C 6 H 5 NH 2
(C 6 H 5) 2 NH
0,78
м-O 2 NC 6 H 4 NH 2
Реакции с С-электрофилами
Важнейшими реакциями этого типа являются алкилирование и ацилирование, которые протекают по атому азота и аналогичны реакциям алифатических аминов. Ароматические амины менее реакционноспособны из-за меньшей основности атома азота.
Реакции ароматического электрофильного замещения
а) Галогенирование
Аминогруппа является сильным активирующим заместителем и ориентантом I рода. Галогенирование свободных аминов протекает очень легко и часто приводит к полигалоидпроизводным. Например, анилин при действии бромной воды мгновенно превращаются в нерастворимое 2,4,6-трибромпроизводное.

Для получения моногалогенпроизводных активирующее действие аминогруппы снижают путем ацилирования. После снятия ацильной защиты путем гидролиза получают свободный амин.


б) Нитрование
При нитровании нитрующей смесью амины окисляются. Кроме того, из-за солеобразования по аминогруппе образуется м-изомер (-NH 3 + - ориентант II рода).

Для введения нитрогруппы в орто - или пара -положение к аминогруппе последнюю защищают ацилированием. Варьируя условия реакций (температуру, нитрующие агенты), можно проводить нитрование региоселективно.

После снятия ацетильной защиты получают свободные орто - и пара -нитроанилины.
в) Сульфирование
Сульфированием ароматических аминов получают аминосульфокислоты. В 90-100%-ной серной кислоте или олеуме амин полностью находится в протонированной форме. Аммониевая группа NH 3 + как сильный электроакцепторный заместитель вызывает резкое замедление реакции сульфирования и ориентирует замещение в мета -положение.

Для получения орто- и пара -аминобензолсульфокислот используют “метод запекания”. Процесс осуществляют при длительном нагревании гидросульфатов ароматических аминов при 100-200 о С в сухом виде или в высококипящих растворителях. При температуре около 100 о С образуется практически чистый орто -изомер (ортаниловая кислота, продукт кинетического контроля), а при 180-200 о С - пара -изомер (сульфаниловая кислота, продукт термодинамического контроля).

Нитрозирование
Первичные ароматические амины с азотистой кислотой образуют относительно устойчивые соли арилдиазония.
ArNH 2 + NaNO 2 + 2HCl ® ArN 2 + Cl - + NaCl + 2H 2 O
Эту реакцию называют диазотированием (см.лек. №43).
Вторичные ароматические амины при нитрозировании дают N-нитрозамины.
ArNHR + NaNO 2 + HCl ® Ar-N(R)-N=O + NaCl + H 2 O
Третичные ариламины дают продукты нитрозирования в пара-положение ароматического кольца.
Заключается в прямом взаимодействии алкилгалогенидов (первичных и вторичных) с аммиаком или аминами. Этот метод является одним из наиболее простых методов получения аминов и солей тетраалкиламмония и был открыт А. Гофманом в 1849 году.
Алкилирование аммиака
Реакции алкилгалогенидов с аммиаком относятся к процессам бимолекулярного нуклеофильного замещения у насыщенного атома углерода, в которых аммиак или амин выполняют роль нуклеофильного агента. Переходное соединения в таких реакциях более полярны, чем исходные, поэтому скорости реакций резко возрастают в более полярных растворителях. В качестве растворителей обычно применяют спирты (этанол или метанол), и более эффективные биполярные апротонные растворители (ДМФА, ДМАА). Реакции алкилирования аммиака приводящие к получению аминов нашли широкое применение как в лабораторной практике, так и в промышленности. Продуктами такого взаимодействия будут смеси первичных, вторичных и третичных аминов, а в случае наличия избытка алкилгалогенида в продуктах реакции также будут находится соли тетраалкиламмония:
Рисунок 1.
Алкиламмониевые катионы обладают свойствами слабых кислот. В результате процесса переноса протонов к молекулам аммиака образуются первичные амины и катионы аммония. Первичные амины проявляют свойства более сильных нуклеофильных агентов, чем исходный аммиак, и при взаимодействии с алкилгалогенидами дают катионы диалкиламмония, из которых далее получаются вторичные амины. Этот процесс может продолжаться далее, приводя к третичным аминам и даже к солям тетраалкиламмония. Вся указанная последовательность происходящих последовательных превращений описывается на приведенной выше схеме (уравнения (1)-(7)). Количественное соотношение первичных, вторичных, третичных аминов и солей тетраалкиламмония зависит от соотношения исходных реагентов.
Увеличение количества алкилгалогeнидов способствует увеличению доли третичных аминов и четвертичных аммониевых солей, в то время как в случае наличия избытка аммиака преимущественно образуются смеси первичных и вторичных аминов. Однако даже в случае наличия большого избытка аммиака реакции невозможно остановить на стадиях образования только первичных аминов. В одном из типичных примеров - реакции одного моля 1-бромоктана и трех молей $NH_3$ при 20$^\circ$С образуется смесь, состоящая из 45% первичного амина - октиламина, 43% вторичного амина - диоктиламина и следов третичного амина - триоктиламина. При больших количествах аммиака доля первичных аминов возрастает, но вторичные амины всегда присутствуют в продуктах реакции.
Рисунок 2.
Таким образом, прямое алкилирование оказывается малоудовлетворительным методом для получения чистых первичных, вторичных и третичных аминов.
Алкилирование аминов
Алкилирование аминов (амино-дегалогенирование) представляет собой тип органической реакции между алкилгалогенидом и амином. Реакция протекает по пути нуклеофильного алифатического замещения (замещения галогенида), и продукт реакции представляет собой более замещенный амин. Метод широко используется в лабораторных условиях, но является менее важным в промышленном отношении, где алкилгалогениды не являются предпочтительными алкилирующими агентами.
Рисунок 3.
В случае, когда в реакции используется третичный амин, продукт реакции представляет собой соль четвертичного аммония в реакции Меншуткина:
Рисунок 4.
Реакция Меншуткина (1890 г.) - особый вид реакции алкилирования алкилгалогенидов третичными аминами, в результате которой образуются четвертичные аммониевые солеи.
Рисунок 5.
Амины и аммиак, как правило, достаточно основные, чтобы участвовать в прямом алкилировании, часто в мягких условиях. Реакции трудно контролировать, так как продукт реакции (первичный амин или вторичный амин), часто являются более нуклеофильными, чем предшественники, и таким образом, также будут вступть в реакцию с алкилирующим агентом. Например, реакция 1-бромооктана с аммиаом или неразветвленными аминами дает в почти равных количествах первичные и вторичные амины. Таким образом, для лабораторных целей, $N$-алкилирование часто ограничивается синтезом третичных аминов. Заметным исключением является реакционная способность альфа-галоуглеродных кислот, которые позволяют синтезировать первичные амины с аммиаком. Внутримолекулярные реакции галоаминов дают циклические азиридины, азетидины и пирролидины.
$N$-алкилирование представляет собой общий путь получения четвертичных аммониевых солей из третичных аминов, так как дальнейшее их алкилирование не представляется возможным.
Примерами реакций $N$-алкилирования с помощью алкилгалогенидов являются реакции получения бензиланилина, 1-бензилиндолила и азетидина. Еще одним особым примером реакции этого типа является реакция дериватизации циклена. Промышленно этилендиамин получают алкилированием аммиака с 1,2-дихлорэтаном.
Алкилирование ариламинов
В обычных условиях арилгалогениды ($ArX$) с алкилатными аминами реагируют неохотно. Реакция обычно требует "активированных" арилгалогенидов, таких, которые содержат сильные электронакцепторные группы, такие, как нитрогруппы в орто- или пара-положениях к атомам галогена.
Рисунок 6.
Для арилированя аминов с неактивированными арилгалогенидами, полезной является реакция Бухвальда-Хартвиаг. В этом процессе, комплексы палладия служат в качестве катализаторов.
Рисунок 7.
Рисунок 8.
Рисунок 9.
2. Ацилирование и алкилирование аминов
Третичные амины отличаются от первичных и вторичных аминов отсутствием способных к замещению атомов водорода, связанных с азотом. Это различие ясно проявляется при действии ацилирующих и алкилирующих средств; из первичных и вторичных аминов при ацилировании обычно получаются замещенные амиды, тогда как третичные амины выделяются в неизмененном состоянии после прибавления воды или водной щелочи. Атомы водорода в аминогруппе первичных и вторичных аминов могут быть замещены в определенных условиях алифатическим или ароматическим радикалом, или же остатками –СONH 2 , -С1, -Вг и -NO 2 . Эти реакции вкратце рассматриваются ниже.
Ацилирование
Способы, применяемые для ацилирования, могут быть в основном разделены на следующие группы: нагревание аминов с кислотами, взаимодействие аминов с хлорангидридами, бромангидридами или ангидридами кислот и реакция аминов со сложными эфирами, или даже с амидами кислот, дающая обычно худшие результаты.
Первый из этих способов состоит в нагревании амина с избытком соответствующей карбоновой кислоты.
Аналогичным путем получаются высшие гомологи ацетанилида. Этот способ часто применяется для идентификации одноосновных кислот. Интересно отметить, что муравьиная кислота значительно легче, чем ее гомологи, превращается в замещенные формамиды по этому способу. Форманилид легко образуется при нагревании 50%-ной водной муравьиной кислоты с анилином.
Для ацетилирования аминов рекомендуется также пользоваться тиоуксусной кислотой. Преимущество этого способа состоит в том, что ацетилирование анилина и его гомологов протекает в этом случае на холоду. Реакция протекает с выделением сероводорода.
Более удобный и распространенный способ получения ацилированных аминов заключается в применении хлорангидридов или ангидридов кислот. Хлорангидрид кислоты реагирует с избытком амина с образованием ацилированного производного и солянокислой соли амина.
Отделение солянокислой соли от ацилированного производного амина основано на их различной растворимости. Обычно реакцию ведут в таком растворителе, в котором соль амина нерастворима. Кроме того, если ацилированный амин нерастворим в воде, солянокислую соль легко удалить промыванием реакционной смеси водой
Если хлорангидрид кислоты сравнительно устойчив к действию воды и холодного водного раствора щелочи, введение ацильной группы может быть осуществлено по способу Шоттена и Баумана. Амин суспендируют в приблизительно 10%-ном водном растворе щелочи и обрабатывают хлорангидридом кислоты, взятым в 1,25-1,5-кратном против теории количестве. При этом реакционную смесь перемешивают или взбалтывают, пока большая часть хлорангидрида не прореагирует. Избыток хлорангидрида разлагают слабым нагреванием реакционной смеси. Образовавшееся трудно растворимое ацильное производное отфильтровывают, промывают водой до полного удаления щелочи и перекристаллизовывают из подходящего растворителя. Важно, чтобы в процессе реакции водный раствор-все время обладал щелочной реакцией. Этот метод с успехом применяется для хлорангидридов ароматических кислот, арилсульфоновых кислот и пирослизевой кислоты. Следует отметить, что сульфонильные производные первичных аминов растворимы в щелочах, а сульфонильные производные вторичных аминов нерастворимы. На этом основан способ распознавания и разделения первичных и вторичных аминов.
К другим способам ацилирования относятся действие хлорангидрида кислоты на эфирный раствор амина с суспендированным в нем углекислым калием, а также ацилирование в пиридине.
Для ацетилирования первичных ароматических аминов в лабораторных условиях целесообразно пользоваться уксусным ангидридом. Реакция между уксусным ангидридом и анилином или его гомологами протекает очень легко и обычно осуществляется прибавлением уксусного ангидрида к смеси амина примерно с 5-кратным по объему количеством воды. При реакции происходит выделение тепла и смесь быстро густеет, вследствие выделения ацетильного производного. Если амин обладает сравнительно высоким молекулярным весом, обычно удобнее перед прибавлением уксусного ангидрида смешивать основание с разбавленной уксусной кислотой. Применение спирта в качестве растворителя при ацетилировании аминов уксусным ангидридом на холоду имеет то преимущество, что избыток ангидрида легко можно удалить 1-2-кратным выпариванием со спиртом. Ацетилирование уксусным ангидридом в водной или спиртовой среде не дает удовлетворительных результатов при первичных ароматических аминах, содержащих в ядре отрицательные заместители.
Вышеупомянутый способ ацетилирования обладает тем преимуществом, что при этом не наблюдается образования диацетильных производных, что имеет место при применении неразбавленного уксусного ангидрида, например, в результате нагревания 10 г анилина с 40 г уксусного ангидрида в течение 1 часа продукт реакции представляет собой смесь, содержащую 10 г диацетиланилина и 5,6 г ацетанилида18. Наличие заместителей, например -СНз, -NO 2 , -С1, -Вг, в о-положении к аминогруппе благоприятствует образованию диацетильных производных. При нагревании о-толуидина с 4-кратным по весу количеством уксусного ангидрида с обратным холодильником получается диацетил-о-толуидин с прекрасным выходом.
Наличие в ядре ароматического амина нитрогруппы и, в меньшей степени, хлора или брома замедляет реакцию ацетилирования при комнатной температуре. Это явление становится особенно заметным при накоплении отрицательных групп в молекуле. При стоянии раствора 2, 4, б-триброманилина в избытке уксусного ангидрида при комнатной температуре в течение 2 недель ацетильное производное не образуется. Присутствие небольшого количества концентрированной серной кислоты очень сильно катализирует процесс ацетилирования, из 1 в 2, 4, 6-триброманилина в 20 г уксусного ангидрида в присутствии двух капель концентрированной серной кислоты при стоянии в течение 10 мин. при комнатной температуре получается чистый 2, 4, 6-трибромацетанилид. Для выделения продукта реакционную смесь выливают в воду.
Наилучший способ получения ацетильных производных низших алкнланилинов состоит в перегонке смеси равных объемов амина и уксусного ангидрида; выше 200° перегоняется ацетильное производное в довольно чистом состоянии. Многие из этих соединений кристаллизуются при охлаждении.
Третичные амины, благодаря своему строению, не способны к образованию амидов при взаимодействии с хлорангидридами или ангидридами кислот. Однако они могут давать с хлорангидридами продукты присоединения, которые обычно разлагаются при действии воды с образованием исходного амина. Например, продукт присоединения 1 моля пиридина к 1 молю хлористого ацетила при действии спирта превращается в солянокислый пиридин и уксусноэтиловый эфир. Хлористый диаллил также образует продукт присоединения к пиридину. Кроме того, описаны соединения, образующиеся при взаимодействии хлористого бензоила и хлористого ацетила с триэтиламином, пиридином, диметиланилином и некоторыми другими третичными аминами. Продукты присоединения триметиламина к арилсульфохлоридам сравнительно стойки к действию воды и дают хлороплатинаты и хлораураты.
Образование производных мочевины
Соли первичных и вторичных аминов с циановой кислотой более или менее легко изомеризуются с образованием замещенных производных мочевины
RNH 2 HCNO --> RNHCONH 2
Эта реакция аналогична превращению циановокислого аммония в мочевину. Следующие примеры иллюстрируют применение этого способа. Первичные и вторичные амины легко реагируют с эфирами изоциановой кислоты с образованием производных мочевины. Обычно для этой цели применяют фенилизоцианат, который нагревают с эквимолекулярным количеством амина в каком-либо не содержащем гидроксила растворителе, например в петролейном эфире.
Совершенно необходимо предохранять реакционную смесь от доступа влаги и применять лишь тщательно обезвоженные растворитель и амин, так как фенилизоцианат реагирует с водой с образованием дифенилмочевины. α-Нафтилизоцианат более удобен в этом отношении, чем фенилизоцианат, так как он менее чувствителен к действию воды и поэтому при реакции образуется меньшее количество нежелательных побочных продуктов.
Фенилизотиоцианат (фенилгорчичное масло) реагирует с аминами аналогичным образом с образованием соответственных производных тиомочевины. Реакция эта осуществляется в тех же условиях, как и с фенилизоцианатом.
Алкилирование первичных и вторичных аминов
Последовательное замещение алкильными группами атомов водорода, находящихся у азота в первичных аминах, ведет к образованию вторичных и третичных аминов. Введение алкильных групп легко достигается действием на амин соответственного галоидного алкила или алкилсульфата. Состав конечного продукта реакции зависит в значительной степени от относительных количеств взятых в реакцию компонентов, а также и от условий опыта, и обычно очень трудно получить при алкилировании только одно из возможных производных амина и поэтому продукт реакции, как правило, представляет собой смесь вторичного и третичного аминов наряду с значительным количеством непрореагировавшего первичного амина, а часто с примесью некоторого количества соли четвертичного аммониевого основания. Получение сложной смеси при применении галоидного алкила является результатом образования при реакции галоидоводорода, который дает соли с находящимися в реакционной смеси аминами. Распределение галоидоводорода между аминами зависит от их относительной основности, их сравнительного количества, а также от растворимости солей аминов в реакционной смеси. При алкилировании ароматических аминов выделяющийся осадок обычно содержит значительное количество соли исходного амина, а в растворе остается алкилированный амин, который вступает в дальнейшую реакцию с галоидным алкилом. Такие затруднения могут быть преодолены, по крайней мере, до известной степени проведением алкилирования в присутствии веществ, способных связывать образующийся галоидозодород, например, углекислой или двууглекислой соли щелочного металла. Для выделения вторичных аминов, наряду с вышеупомянутыми способами, обычно, за исключением некоторых особых случаев, пользуются способностью вторичных аминов образовывать нитрозамины. При восстановлении нитрозаминов оловом с соляной кислотой или при нагревании их с минеральными кислотами получаются чистые вторичные амины. Другой способ, по которому удается получать вторичные амины с значительно лучшими выходами, основан на способности металлических производных многих замещенных амидов типа RCONHR реагировать с галоидными алкилами. Из продукта алкилирования при гидролизе получается вторичный амин
Для этой цели удобно пользоваться ацетанилидом и его гомологами. Кроме того, применялись и формильные производные первичных ароматических аминов, а также арилсульфонильные производные первичных аминов.
Другой способ получения гомологов метиланилина заключается в нагревании галоидного алкила с большим избытком ароматического амина. По окончании реакции избыток ароматического амина осаждают прибавлением водного раствора хлористого цинка. Этот способ применялся для получения многих алкиланилинов с вполне удовлетворительными результатами. Таким же путем могут быть получены алкиланилины, содержащие третичную алкильную группу. Для алкилирования аминов также можно пользоваться ди-алкилсульфатами. Впрочем, обычно, этот способ ограничивается применением имеющегося в продаже диметилсульфата. Алкилирование по этому способу проводится в индиферентном растворителе или в присутствии водной щелочи, причем последняя модификация имеет более широкое применение. Вместо диалкилсульфатов можно1 применять эфиры арилсульфоновых кислот. Спирты вступают в реакцию с солями первичных ароматических аминов, примерно, при 200° с образованием моно- и диалкилариламинов. Эта реакция имеет применение в промышленности; для получения метиланилина нагревают при 180° смесь 55 частей солянокислого анилина и 16 частей метилового спирта. Для получения диметиланилина смесь 80 частей анилина, 78 частей метилового спирта и 8 частей серной кислоты нагревают в автоклаве до 235°. В лабораторных условиях можно вместо серной кислоты пользоваться другим катализатором, например йодом. Еще более активным катализатором в этой реакции является смесь порошкообразной меди с бромистым натрием или смесь галоидных солей меди и натрия Вторичные амины могут быть также получены восстановлением. Эта реакция может быть осуществлена электролитическим путем, действием цинковой пыли и водной щелочи натрия в спиртовой среде или муравьиной кислоте.
Новый способ получения метальных производных α- и β-нафтиламинов предложен Родионовым и Введенским. Для получения моно- и диметильных производных применяется действие метилового эфира р-толуолсульфоновой кислоты на соответствующий амин.
Другой интересный способ получения вторичных аминов основан на взаимодействии азометинов с йодистыми алкилами, причем образуются соединения, которые по прибавлении воды или спирта расщепляются на вторичный амин и альдегид.
Арилирование первичных и вторичных аминов
Введение в аминогруппу ароматического остатка обычно сопряжено с некоторыми затруднениями, вследствие малой реакционноспособности галоида в ароматических соединениях. Например, хлорбензол и бромбензол не вступают в реакцию с анилином в условиях, аналогичных применяемым для получения этиланилина. Впрочем, в присутствии медной бронзы или йодистой меди эта реакция протекает более гладко.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме.
R""R"R"N + RHal -> R""R"R"RNHal
Реакция образования солей четвертичных аммониевых оснований часто применяется для идентификации третичных аминов, причем в качестве реактива наибольшее применение имеет йодистый метил. Рекомендуется также пользоваться для этой цели метиловым эфиром р-толуолсульфоновой кислоты. Ниже приводятся общие условия реакции для получения р-толуол-сульфоновокислых солей четвертичных аммониевых оснований.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме
R""R"R"N + RHal -> R""R"R"RNHal
Вместо галоидных алкилов можно пользоваться диалкилсульфатами или алкиловыми эфирами ароматических сульфоновых кислот, причем получаются сернокислые или арилсульфоновокислые соли соответствующих четвертичных аммониевых оснований.
Соли четвертичных аммониевых оснований образуются не только в результате взаимодействия галоидных алкилов или эфиров ароматических сульфоновых кислот с третичными амидами, но и при действии эфиров йодуксусной кислоты на некоторые амины. Легче других вступают в эту реакцию бензилпиперидин, алифатические третичные амины и хинолин. В некоторых случаях для получения четвертичных аммониевых солей.
Легкость образования четвертичных аммониевых солеи сильно зависит от характера исходных соединений.
Наличие заместителей в о-положении к аминогруппе оказывает замедляющее действие на скорость реакции, что ясно видно из сравнения констант, для диметил-о-, -т- и р-толуидина, а также для хинолина и изохинолина. Это явление еще более резко выражено при наличии двух заместителей в о-положении к аминогруппе.
Например, третичные амины (III) и (IV) не реагируют с йодистым метилом при 100°, тогда как изомерные им амины, обладающие другим строением, сравнительно легко образуют четвертичные аммониевые соли

Диметилмезидин (V) и диметиламинопентаметилбензол (VI) также не способны к образованию четвертичных аммониевых соединений
N-метилакридиния), и осадок (5-окси-N-метилакридан) окисляют хромовым ангидридом. Акридон сульфируется и нитруется в положения 3 и 3,7, а при бромировании дает 2,3-дибромпроизводное. Мною апробирован метод получения акридона из фенилантраниловой кислоты. Выбранная реакция принадлежит к реакциям замыкания цикла. ОБЗОР ЛИТЕРАТУРЫ Реакции замыкания цикла. Типы реакций. Реакции замыкания...





Получаемый ПВХ отличается высокой полидисперсностыо и широким молекулярно-массовым распределением. Достоинства полимеризации в массе: высокая чистота полимера, его повышенные электроизоляционные свойства, прозрачность изделий. Производство поливинилхлорида в суспензии Большая часть ПВХ производится суспензионным методом, обеспечивающим высокое качество полимера (со сравнительно узким...

И, конечно же, за многими другими, которые будут получены, - будущее. В этом направлении и работают многие НИИ и исследователи. Аспекты поиска новых лекарств, изыскание новых лекарственных веществ состоит из трех основных этапов: химический синтез, установление фармакологической активности и безвредности (токсичности). Такая стратегия поиска с большой затратой времени, реактивов, животных, труда...
Ароматические амины способны замещать водород аминогруппы на алкилы. Эта реакция приводит к вторичным и третичным аминам:
C 6 H 5 NH 2 + CH 3 I → C 6 H 5 -NH-CH 3 + CH 3 I → C 6 H 5 -N(CH 3) 2
Алкилированиеведут спиртами или хлоралаканами, в качестве катализаторов используют соли одновалентной меди в виде аммиачных комплексов. Важно, что процесс алкилирования является последовательно-параллельным. Это обусловлено тем, что образовавшийся амин, в свою очередь, способен реагировать с алкилирующим агентом. Состав продуктов зависит от соотношения реагентов.
2. Ацилирование ароматических аминов
При действии ацилирующих агентов (кислоты, ангидриды, хлорангидриды) водородные атомы аминогруппы замещаются на ацильные остатки.
Ацильные производные не обладают основными свойствами. Они обладают устойчивостью к окислителям и потому используются в качестве промежуточных веществ в реакциях аминов в присутствии окислителей (например, нитрование).
3. Синтез азометинов (оснований Шиффа)
При слабом нагревании ароматических первичных аминов с ароматическими альдегидами легко образуются так называемые основания Шиффа или азометины:

Под действием разбавленных кислот основания Шиффа гидролизуются до альдегида и амина.
4. Реакции аминов с азотистой кислотой
Первичные ароматические амины с азотистой кислотой при 0 – 5°С образуют соли диазония:

Вторичные амины при взаимодействии с азотистой кислотой образуют N-нитрозо-N-метиланилины:

Третичные амины с азотистой кислотой вступают в реакцию электрофильного замещения:

Важнейшие представители ароматических аминов
Анилин впервые был получен в результате перегонки индиго с известью (1826 г.). В 1842 г. его получил Зинин восстановлением нитробензола. В незначительных количествах содержится в каменноугольной смоле. В промышленности получают из нитробензола каталитическим гидрированием с медным катализатором в газовой фазе. Анилин в больших количествах идет на получение красителей, циклогексиламина, капролактама, пестицидов и др.
п-Толуидин широко применяется в производстве красителей, особенно фуксина.
N , N -диметиланилин применяется в производстве красителей и взрывчатых веществ.
Лекция 28. ДИАЗОСОЕДИНЕНИЯ. АЗОСОЕДИНЕНИЯ
Реакция диазотирования, условия проведения, механизм. Влияние заместителей в бензольном кольце на скорость реакции. Строение диазосоеиинений в зависимости от рН, таутомерные превращения. Химические свойства. Реакции, протекающие с выделением азота: нуклеофильное замещение диазониевой группы на гидроксил, алкоксигруппу, галогены. Механизм реакции. Реакции, протекающие без выделения азота. Условия реакции азосочетания с аминами и фенолами. Влияние заместителей на реакционную способность диазосоединения. Понятие об азокрасителях.
Реакция первичных ароматических аминов с азотистой кислотой приводит к образованию солей диазония (II. Гриси, 1858 г.). Эти соли имеют общую формулу [Аг-N≡N] + X - (где Х Cl, Br, NO 2 , HSO 4 , и т.д.):

Названия солей диазония образуются добавлением окончания -диазоний к названию радикала исходного ароматического соединения с указанием названия аниона, например фенилдиазоний хлорид или хлористый фенилдиазоний.